


Genetica dei disturbi della crescita e dello sviluppo sessuale
La crescita di ogni bambino è influenzata da diversi fattori: la nutrizione materna e l'ambiente intrauterino influenzano la crescita al momento della nascita e durante il primo mese di vita; i fattori genetici ed il tipo di alimentazione influenzano la crescita successivamente. In una data popolazione sono stabiliti parametri di normalità relativi ad altezza, peso e circonferenza della testa che devono essere misurati costantemente nel tempo. Se un bambino cresce regolarmente, questo suggerisce uno stato di buona salute, mentre una crescita più lenta del normale aumenta la possibilità che vi possa essere una malattia cronica sottostante oppure una causa endocrinologica.
Normalmente, i bambini crescono molto nei primi due anni di vita, poi la loro crescita rallenta fino a quando arrivano alla pubertà.


Ritardi e difetti di crescita
Si definisce ritardo di crescita un ritardo dell’aumento di peso e della crescita fisica che può portare a ritardo di sviluppo e maturazione.
Il ritardo di crescita si manifesta se un bambino è costantemente sottopeso, in genere sotto il 3°-5° percentile, rispetto ai coetanei dello stesso sesso, ma soprattutto quando il bambino cresce con una velocità (calcolata come cm/anno) inferiore al 25° percentile per sesso ed età. Le cause di un ritardo di crescita possono essere molteplici. La Società Europea di Endocrinologia Pediatrica classifica il ritardo di crescita in: disordine primario, disordine secondario e bassa statura idiopatica.
Tra i disordini primari della crescita è presente il ritardo di crescita sindromico associato ad anomalie cromosomiche o a difetti monogenici, che interessano geni a localizzazione ossea (deficit del gene SHOX, ipocondroplasia/acondroplasia, osteogenesi imperfetta) e geni che interessano direttamente o indirettamente la secrezione degli ormoni deputati alla crescita (ad es. gene del GH, del GHRH, dell’IGF-1 e fattori di trascrizione implicati nell’embriogenesi della regione diencefalo-ipofisaria).
Tra i disordini secondari della crescita rivestono un ruolo molto importante alcune patologie croniche (cardiache, respiratorie, epatiche, renali, ematologiche e intestinali da malassorbimento come la celiachia e le malattie infiammatorie intestinali); meno frequenti, ma di fondamentale importanza sono invece le endocrinopatie non congenite (disordini secondari dell’asse GH-IGF-1, ipotiroidismo, ipercortisolismo, pseudoipoparatiroidismo).
È quindi fondamentale definire il ritardo di crescita in modo da scegliere l’esame appropriato.
Il rapido sviluppo tecnologico ha portato nuove scoperte sulle cause genetiche dei disturbi congeniti, comprese le forme sindromiche e non sindromiche di difetti della crescita; in particolare la combinazione di citogenetica classica (Cariotipo), con la Citogenomica (CGH/SNIP-array) e il Sequenziamento di Nuova Generazione (NGS), ha portato ad una migliore comprensione delle cause genetiche dei disturbi della crescita.
Cariotipo
Nei casi di disturbi dell’accrescimento sindromico è fondamentale eseguire l’analisi del cariotipo che permette di studiare il numero e la struttura dei cromosomi. Tale tecnica è in grado di individuare l’assenza di un cromosoma o la presenza di un cromosoma sovra-numerario che potrebbero essere causa di ritardo di crescita (ad es. Sindrome di Turner 45,X; Sindrome di Down 47,XX+21).
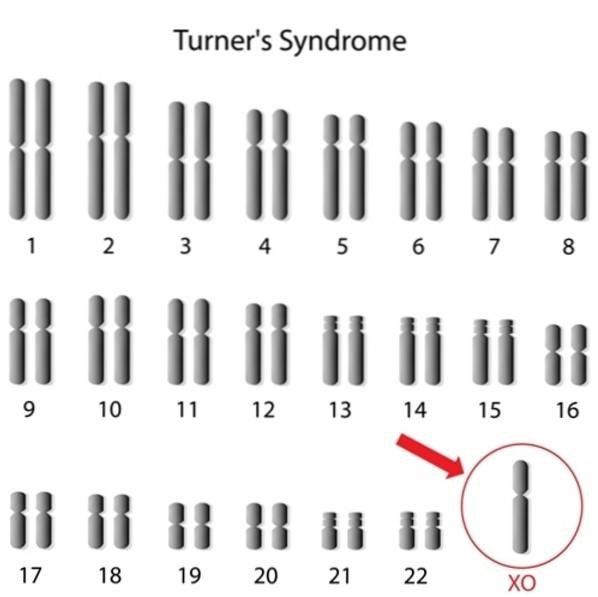
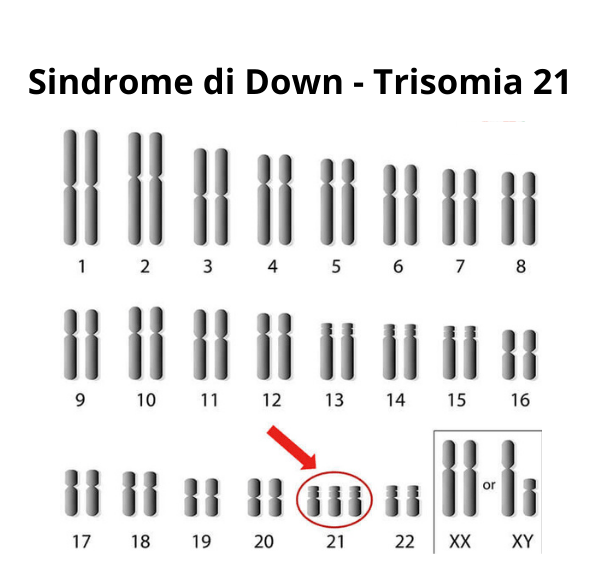
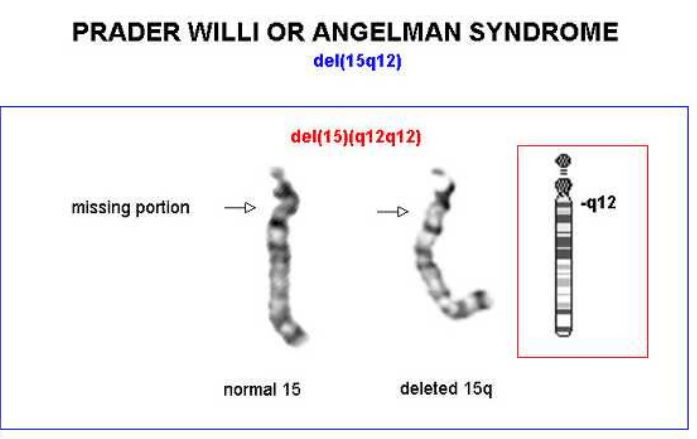
Il cariotipo, in combinazione all’ibridazione fluorescente in situ (FISH), ci permette di individuare variazioni strutturali dei cromosomi, delezioni o duplicazioni, che potrebbero essere causa di disturbi dell’accrescimento, come ad es. la sindrome di Prader-Willi, causata nel 70-80% dei casi, dalla perdita di un pezzetto del cromosoma 15 di origine paterna.
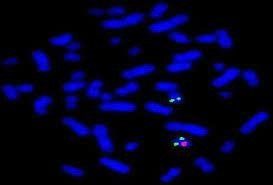
La tecnica
Campione richiesto: sangue in eparina
Tempo di refertazione: 25 giorni
Cosa si analizza: Il corredo cromosomico dell’individuo
Metodo analitico: Coltura cellulare linfocitaria
Consulenza genetica: Fornita dallo specialista in Genetica prima dell’esecuzione dell’esame e alla consegna del referto per l’interpretazione del risultato
CGH ARRAY
L’array-CGH viene prevalentemente utilizzato per la diagnosi postnatale di fenotipi complessi associati a ritardo mentale di grado variabile. Sono numerose le CNVs che concorrono alla clinica di importanti disordini dello sviluppo, in particolare i disturbi generalizzati dello sviluppo quali i disordini dello spettro autistico, le disabilità intellettive di grado variabile, diverse forme di epilessia, la schizofrenia e disturbi della crescita di natura variabile.
È stato stimato che in Italia ogni anno mediamente 1000 persone con deficit intellettivo o difetti congeniti resterebbero privi di una diagnosi se non venisse utilizzata l’indagine in Array come test diagnostico. Il test eseguito in epoca pediatrica fornisce informazioni utili ai genitori per capire le future esigenze del bambino, per dare la possibilità di accedere a particolari servizi e gruppi associativi di supporto e per aiutarli nel futuro processo decisionale riproduttivo. In più, offre ai genitori la possibilità di uscire da un senso di frustrazione legato alla non conoscenza delle cause del problema e del rischio ad esso associato.
La tecnica
Campione richiesto: sangue in EDTA
Geni analizzati: tutto il genoma ad una risoluzione di 75 Kb (piattaforma Array 180K)
Metodo analitico: Array CGH
Consulenza genetica: fornita dallo specialista in Genetica prima dell’esecuzione dell’esame e alla consegna del referto per l’interpretazione del risultato.
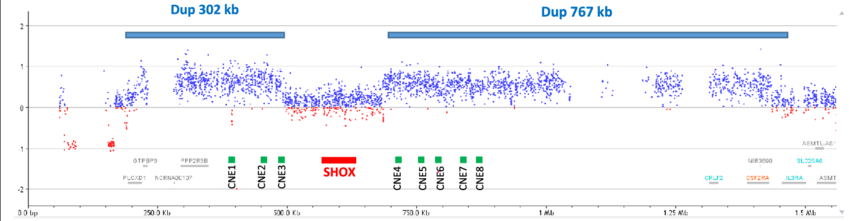
Pannello ritardo di crescita
Le cause del ritardo di crescita possono essere di origine genetica, dovute a mutazioni puntiformi o piccole delezioni/duplicazioni rilevabili mediante sequenziamento del DNA basato su PCR. I geni interessati sono molteplici, ad esempio la condizione di deficit isolato di GH (IGHD) può essere dovuta ad una o più mutazioni geniche che interferiscono direttamente sulla sintesi dell’ormone della crescita o sulla trasmissione del segnale. Anche mutazioni nel recettore per l’ormone della crescita (GHR) possono comportare un fenotipo molto simile a quello di un IGHD, come per esempio nella sindrome di Laron, dove mutazioni nel GHR causano resistenza completa al trattamento con GH.
La Biogenet ha sviluppato un pannello NGS dedicato al “Ritardo di crescita”.
Il pannello comprende 36 geni associati a diverse manifestazioni cliniche correlate a deficit della crescita: bassa statura da difetto quantitativo dell'ormone della crescita; deficit isolato dell'ormone della crescita (tipo 1A, 1B, tipo 2 e legato all’X); bassa statura da deficit parziale di GHR o di GHSR e/o da deficit combinati dell'ormone ipofisario; ritardo di crescita sindromico (Sindrome di Laron, Sindrome 3M, CHARGE, Kallmann, Omenn, Sindrome di Silver-Russell da mutazione puntiforme); Ipogonadismo ipogonadotropo congenito normosmico; bassa statura SHOX-correlata; ritardo della crescita da deficit del fattore di crescita 1 insulino-simile o da deficit primitivo della subunità acido-labile; ipotiroidismo da deficit dei fattori di trascrizione implicati nello sviluppo o nella funzione ipofisaria.
Qui LINK è possibile conoscere i geni che vengono indagati nel pannello NGS – Ritardo di Crescita da noi formulato.
Pannello Ipogonadismo Ipogonadotropo
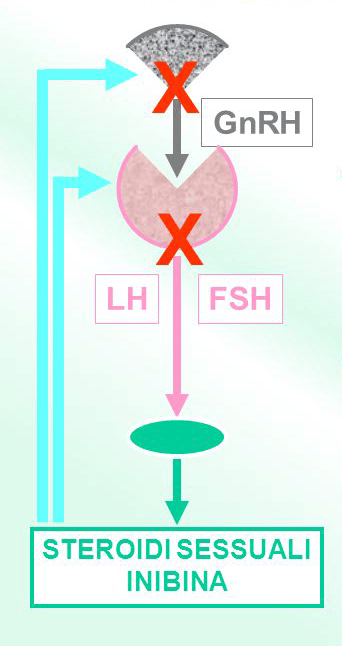
La pubertà è un periodo di significativi cambiamenti emotivi e fisici del corpo, si verifica normalmente tra gli 8 e i 13 anni per le femmine e tra i 9 e i 14 anni per i maschi. L’assenza completa o parziale di pubertà è diagnosticata tipicamente negli adolescenti ed è caratterizzata da bassi livelli di steroidi sessuali estrogeni e progesterone (ipogonadismo), causato dal mancato rilascio di tali ormoni mediato dal GnRH (ormone di rilascio delle gonadotropine), Ipogonadismo Ipogonadotropo [HH]. Ci sono due percorsi biologici che possono portare ad ipogonadismo ipogonadotropo. In un primo caso i neuroni secernenti GnRH migrano dal bulbo olfattivo all'ipotalamo durante lo sviluppo embriologico. Le mutazioni che interferiscono con questa migrazione portano all'HH anosmico, noto anche come sindrome di Kallmann. La seconda via comporta mutazioni con perdita di funzione che determinano un malfunzionamento dei neuroni secernenti GnRH che hanno subito la normale migrazione embrionale. Queste mutazioni causano ipogonadismo ipogonadotropo normosmico (nHH).
La Biogenet ha sviluppato un pannello NGS dedicato all' “ Ipogonadismo Ipogonadotropo”.
Il nostro pannello NGS indaga 26 geni, noti per essere correlati a questo disturbo.
Da questo LINK è possibile conoscere i geni che vengono indagati nel pannello NGS – Ipogonadismo Ipogonadotropo da noi formulato
La tecnica
Campione richiesto: sangue in EDTA
Metodo analitico: estrazione del DNA, Arricchimento selettivo delle sequenze dei geni d’interesse, Sequenziamento di ultima generazione (NGS)
Consulenza genetica: fornita dallo specialista in Genetica prima dell’esecuzione dell’esame e alla consegna del referto per l’interpretazione del risultato.

INDIRIZZO
P.IVA 02229720780
CONTATTI
ORARI DELLO STUDIO
- Lun - Ven
- - -
- Sabato
- -
- Domenica
- Chiuso
 |
Questa azienda è presente anche su
|
Questa azienda è presente anche su  e
e 